From quantum mechanics to computer calculation
Computational quantum chemistry aims to solve the electronic Schrödinger equation for real molecules. The time-independent Schrödinger equation reads:
Ĥ ψ = E ψ
where Ĥ contains electronic kinetic energy, electron–electron repulsion, and nucleus–electron attraction terms. The problem is exactly solvable for H and H₂⁺; for every other system, systematic approximations are required.
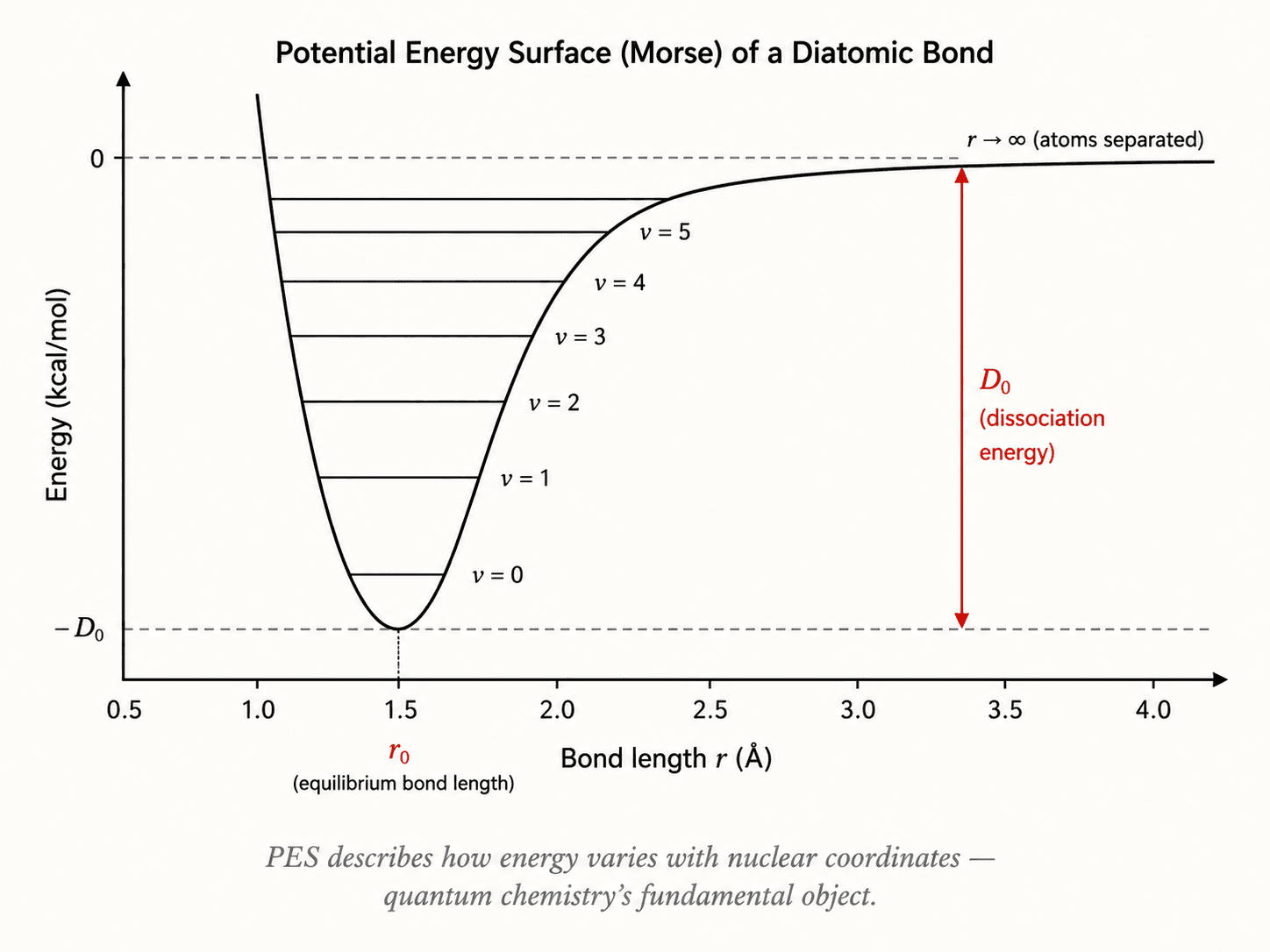
The Born-Oppenheimer approximation is the first: nuclei, being far heavier, are treated as fixed while electrons redistribute. This yields a potential energy surface (PES) as a function of nuclear coordinates.
Hartree-Fock: the single-determinant limit
The Hartree-Fock (HF) method represents the many-electron wavefunction by a single Slater determinant:
ψ_HF = (1/√N!) det[χ₁χ₂…χ_N]
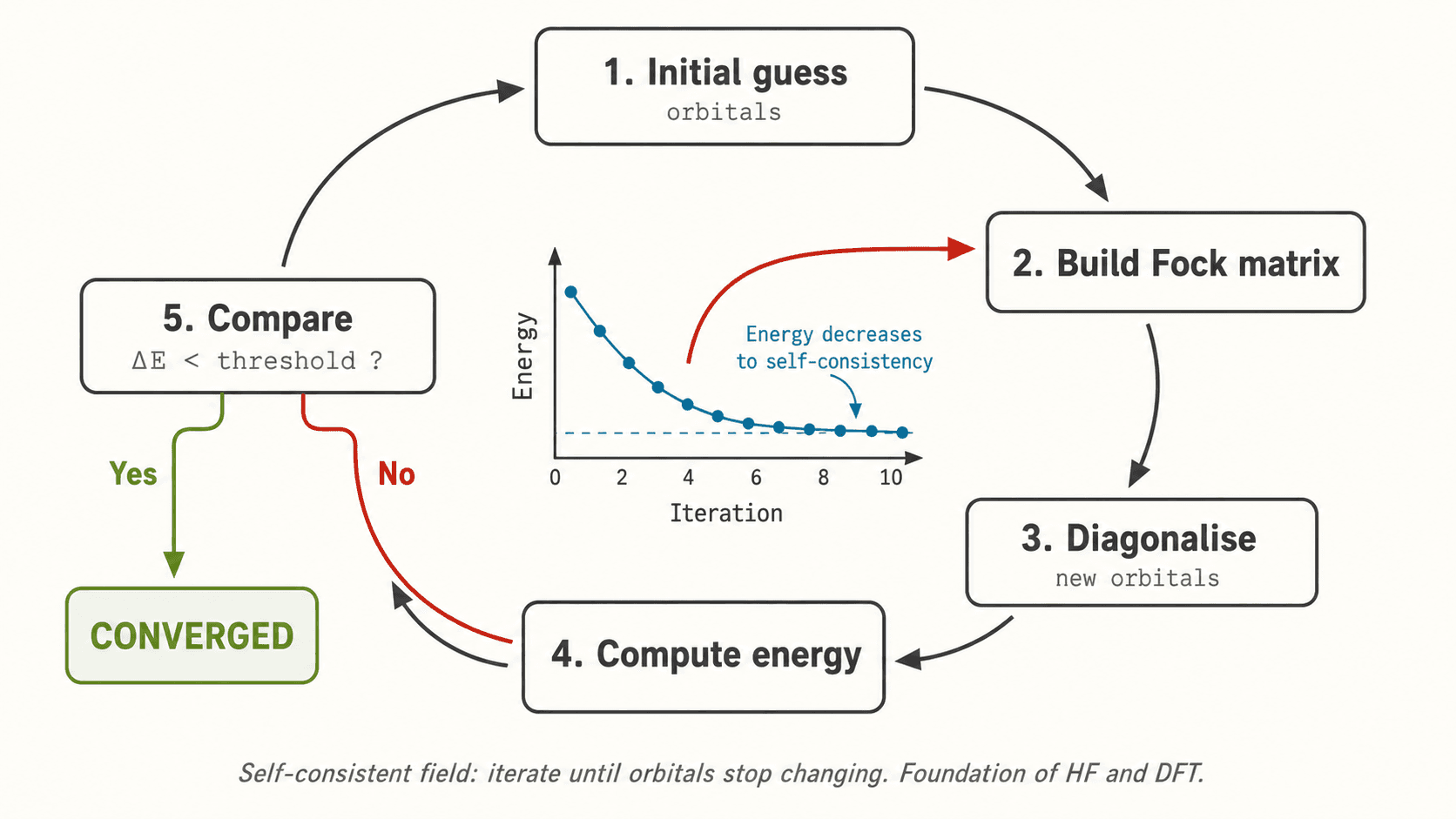
where χ_i are spin-orbitals. Each electron experiences the mean field of all others (self-consistent field, SCF). The HF energy is the best achievable with one determinant.
The energy missing relative to the exact non-relativistic value is the correlation energy:
E_corr = E_exact − E_HF (< 0)
It represents roughly 1 % of total energy yet can account for 100 % of a reaction energy. Post-HF methods (MP2, CCSD, CCSD(T)) recover this correlation at increasing computational cost (O(N⁵) to O(N⁷)).
Density Functional Theory (DFT)
DFT rests on the Hohenberg-Kohn theorems (1964): 1. The ground-state energy is a unique functional of the electron density ρ(r). 2. The variational principle ensures the true density minimises the energy.
The DFT energy is written:
E[ρ] = T[ρ] + V_ee[ρ] + V_ext[ρ]
The problematic term is the exchange-correlation energy E_xc[ρ], whose exact form is unknown. Kohn and Sham (1965) reformulated the problem by introducing a fictitious non-interacting electron system with the same density as the real one, reducing DFT to a tractable orbital problem.
| Functional family | Examples | Strengths | Weaknesses |
|---|---|---|---|
| LDA (local density) | VWN, PW92 | Metallic solids | Overbinds |
| GGA (generalised gradient) | PBE, BLYP | Organic molecules | No dispersion |
| Hybrids | B3LYP, PBE0 | Geometries, frequencies | ×2–4 cost vs GGA |
| Range-separated hybrids | ωB97X-D | Charge transfer | More empiricism |
| Meta-GGA | TPSS, M06-L | Thermochemistry | Numerical instability |
B3LYP is historically the most cited functional in organic chemistry; PBE dominates for solids and physics.
Basis sets: the language of calculation
Kohn-Sham orbitals are expanded in a basis set — a set of known functions. Two main families exist:
Gaussian-type functions (GTF): φ(r) = N · r^l · exp(−αr²). Two-electron integrals are evaluated analytically. Pople bases (6-31G, 6-311+G*), Dunning bases (cc-pVDZ, aug-cc-pVTZ), Ahlrichs bases (def2-SVP, def2-TZVP).
Plane waves: used in solid-state physics with pseudopotentials; not suited to isolated molecules.
The star notation (one or two asterisks) indicates polarisation functions (d on heavy atoms, p on H). Diffuse functions (prefixed with + or aug-) are essential for anions and excited states.
| Basis set | Size | Typical use |
|---|---|---|
| 6-31G* | Small | Preliminary optimisation |
| 6-311+G** | Medium | Reliable thermochemistry |
| def2-TZVP | Medium-large | Current standard |
| aug-cc-pVTZ | Large | Spectroscopy, anions |
Reading a Gaussian output file
A typical Gaussian calculation (B3LYP/6-31G* on water) produces structured output:
1. Header: charge, multiplicity, basis, functional, version. 2. Initial geometry: coordinates in Ångströms (Z-matrix or Cartesian). 3. SCF cycles: convergence of energy (~10⁻⁸ hartree) and density. 4. HF/DFT energy: in hartree (1 Eh = 2625.5 kJ/mol). 5. Optimised geometry: if opt is requested — bond lengths (Å), angles (°). 6. Harmonic frequencies: if freq — values in cm⁻¹ (negative = transition structure). 7. NBO/Mulliken charges: electron density analysis.
"Essentially, all models are wrong, but some are useful." — George Box. In DFT, B3LYP/6-31G* remains useful to explore trends, even though def2-TZVP/PBE0-D3 is preferable for publication.
The key quantity for comparing molecules is the thermochemical Gibbs energy G = E_elec + ZPE + H_therm − TS, accessible with the freq=temperature keyword.
Limitations and outlook
Standard DFT underestimates van der Waals (London) dispersion. Grimme's empirical corrections (D3, D3BJ) or the non-local VV10 functional remedy this. Multi-reference transition-metal states (Fe, Mn in enzymes) remain difficult for DFT; CASSCF/NEVPT2 is required. DFT-scale calculations for thousands of atoms are possible with DFTB (approximate tight-binding) or QM/MM methods.