De la mécanique quantique au calcul sur ordinateur
La chimie quantique computationnelle ambitionne de résoudre l'équation de Schrödinger électronique pour des molécules réelles. L'équation de Schrödinger indépendante du temps s'écrit :
Ĥ ψ = E ψ
où Ĥ contient les termes d'énergie cinétique électronique, la répulsion électron–électron et l'attraction noyau–électron. Le problème est exactement soluble pour H et H₂⁺ ; pour tout autre système, on recourt à des approximations systématiques.
L'approximation de Born-Oppenheimer est la première : les noyaux, beaucoup plus lourds, sont traités comme fixes pendant que les électrons se redistribuent. On obtient ainsi une surface d'énergie potentielle (PES) en fonction des coordonnées nucléaires.
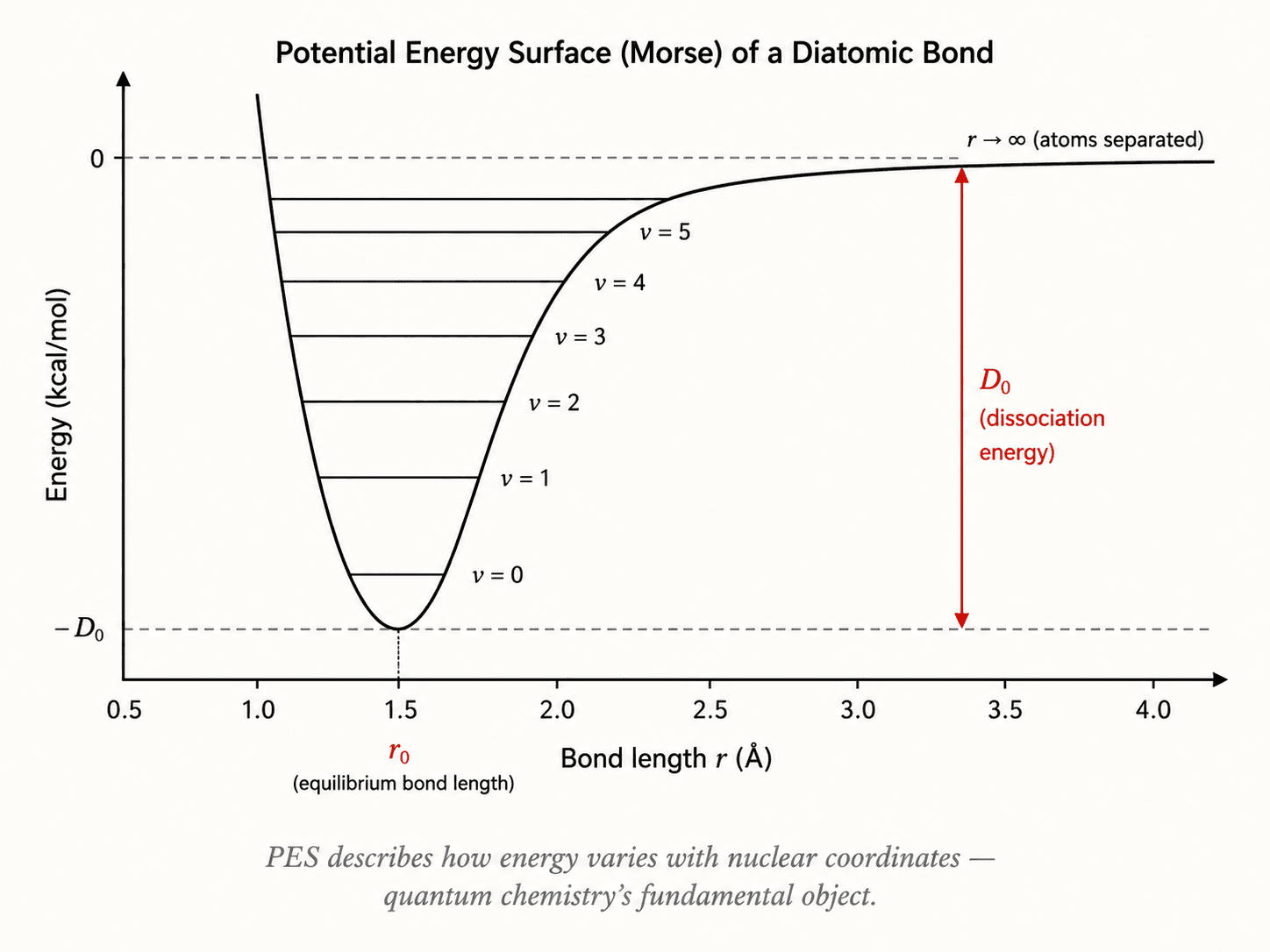
Hartree-Fock : la limite d'un déterminant
La méthode Hartree-Fock (HF) représente la fonction d'onde multiélectronique par un seul déterminant de Slater :
ψ_HF = (1/√N!) det[χ₁χ₂…χ_N]
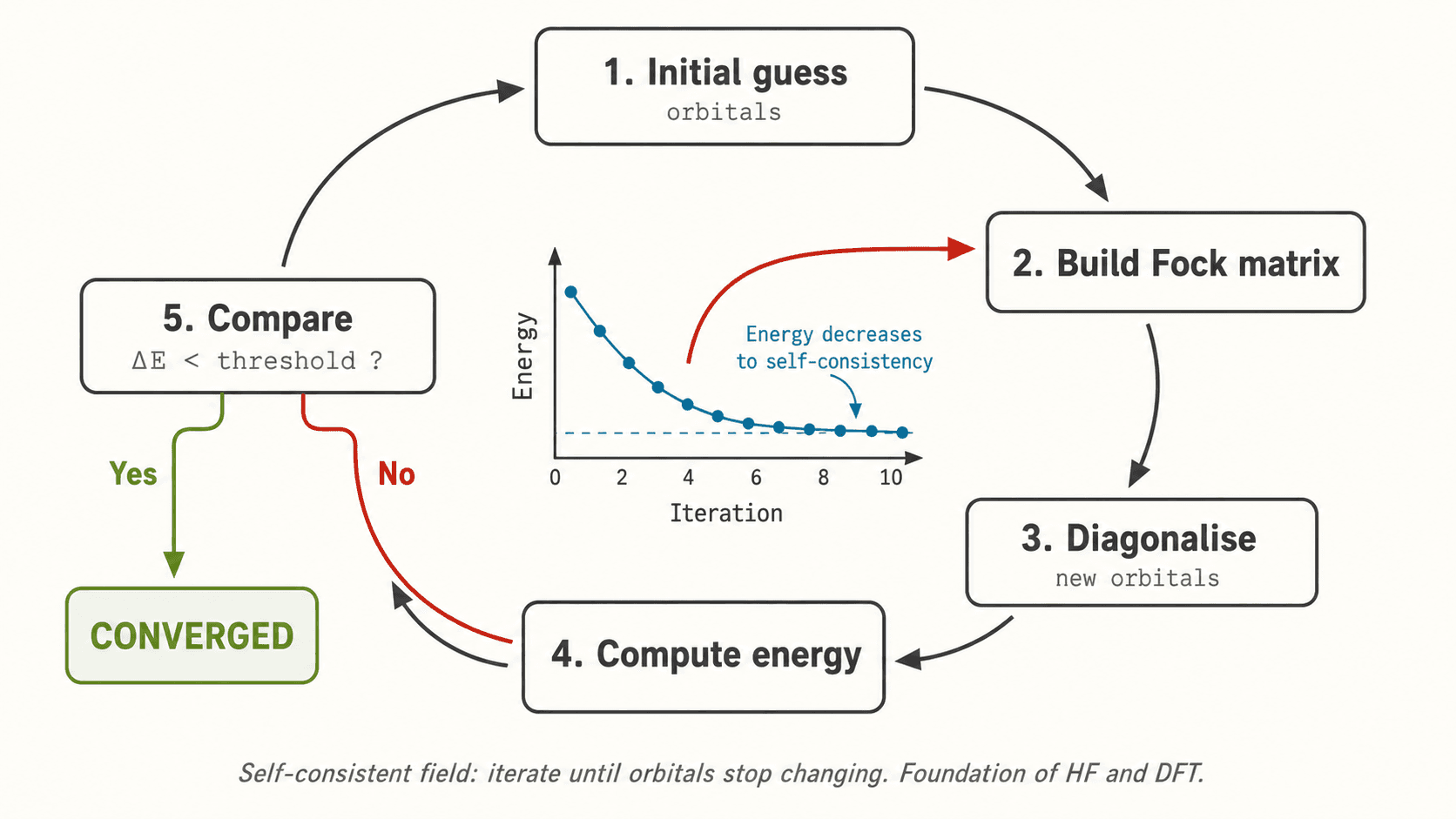
où les χ_i sont des spin-orbitales. Chaque électron ressent le champ moyen des autres (approximation du champ self-consistant, SCF). L'énergie HF est la meilleure énergie atteignable avec un seul déterminant.
L'énergie manquante par rapport à la valeur exacte non-relativiste s'appelle énergie de corrélation :
E_corr = E_exact − E_HF (< 0)
Elle représente environ 1 % de l'énergie totale mais peut atteindre 100 % de l'énergie de réaction. Les méthodes post-HF (MP2, CCSD, CCSD(T)) récupèrent cette corrélation au prix d'un coût calcul élevé (O(N⁵) à O(N⁷)).
La théorie de la fonctionnelle de la densité (DFT)
La DFT repose sur les théorèmes de Hohenberg-Kohn (1964) : 1. L'énergie fondamentale est une fonctionnelle unique de la densité électronique ρ(r). 2. Le principe variationnel assure que la vraie densité minimise l'énergie.
L'énergie DFT s'écrit :
E[ρ] = T[ρ] + V_ee[ρ] + V_ext[ρ]
Le terme problématique est l'énergie d'échange-corrélation E_xc[ρ], dont la forme exacte est inconnue. Kohn et Sham (1965) reformulent le problème en introduisant un système fictif d'électrons non interagissants ayant la même densité que le système réel, ramenant la DFT à un problème orbital tractable.
| Famille de fonctionnelles | Exemples | Forces | Limites |
|---|---|---|---|
| LDA (densité locale) | VWN, PW92 | Solides métalliques | Surestime les énergies de liaison |
| GGA (gradient généralisé) | PBE, BLYP | Molécules organiques | Dispersion absente |
| Hybrides | B3LYP, PBE0 | Géométries, fréquences | Coût ×2–4 vs GGA |
| Hybrides à séparation de plage | ωB97X-D | Transfert de charge | Plus d'empirisme |
| Meta-GGA | TPSS, M06-L | Thermochimie | Instabilité numérique |
La fonctionnelle B3LYP est historiquement la plus citée en chimie organique ; PBE domine pour les solides et la physique.
Bases d'orbitales : langages du calcul
Les orbitales de Kohn-Sham sont développées sur un ensemble de fonctions de base (basis set). On distingue deux grandes familles :
Fonctions de type gaussiennes (GTF) : φ(r) = N · r^l · exp(−αr²). Les intégrales biélectroniques se calculent analytiquement. Bases de Pople (6-31G, 6-311+G*), bases de Dunning (cc-pVDZ, aug-cc-pVTZ), bases Ahlrichs (def2-SVP, def2-TZVP).
Ondes planes : utilisées en physique du solide avec des pseudopotentiels ; non adaptées aux molécules isolées.
La notation étoilée (un ou deux astérisques) indique l'ajout de fonctions de polarisation (d sur les atomes lourds, p sur H). Les fonctions diffuses (préfixées par + ou aug-) sont indispensables pour les anions et les états excités.
| Base | Taille | Usage typique |
|---|---|---|
| 6-31G* | Petite | Optimisation préliminaire |
| 6-311+G** | Moyenne | Thermochimie fiable |
| def2-TZVP | Moyenne-large | Standard actuel |
| aug-cc-pVTZ | Grande | Spectroscopie, anions |
Lire un fichier de sortie Gaussian
Un calcul Gaussian typique (B3LYP/6-31G* sur l'eau) produit une sortie structurée :
1. En-tête : charge, multiplicité, base, fonctionnelle, version. 2. Géométrie initiale : coordonnées en Ångströms (Z-matrix ou cartésiennes). 3. Cycles SCF : convergence de l'énergie (seuil ~10⁻⁸ hartree) et de la densité. 4. Énergie HF/DFT : en hartree (1 Eh = 2625,5 kJ/mol). 5. Géométrie optimisée : si opt est demandé — longueurs de liaisons (Å), angles (°). 6. Fréquences harmoniques : si freq — valeurs en cm⁻¹ (négatives = structure de transition). 7. Charges NBO/Mulliken : analyse de la densité de charge.
"Essentially, all models are wrong, but some are useful." — George Box. En DFT, B3LYP/6-31G* reste utile pour explorer des tendances, même si def2-TZVP/PBE0-D3 est préférable pour la publication.
La grandeur clé pour comparer des molécules est l'énergie de Gibbs thermochimique G = E_elec + ZPE + H_therm − TS, accessible avec le mot-clé freq=temperature.
Limites et perspectives
La DFT standard sous-estime les interactions de van der Waals (London). Les corrections empiriques de Grimme (D3, D3BJ) ou la fonctionnelle non-locale VV10 y remédient. Les états de transition métalliques à multi-référence (Fe, Mn dans les enzymes) restent difficiles pour la DFT ; on recourt à CASSCF/NEVPT2. Le calcul à l'échelle DFT pour des systèmes de milliers d'atomes est possible avec DFTB (tight-binding approché) ou les méthodes QM/MM.