Principe physique de la RMN
La Résonance Magnétique Nucléaire (RMN) repose sur le moment magnétique de spin nucléaire. Les noyaux de spin I ≠ 0 (¹H, ¹³C, ¹⁹F, ³¹P…) se comportent comme de petits aimants. Placés dans un champ magnétique statique B₀, leurs spins se distribuent en 2I+1 niveaux d'énergie ; pour I = 1/2 (¹H, ¹³C) il y a deux niveaux α (parallèle, énergie basse) et β (antiparallèle).
La différence d'énergie ΔE = h·γ·B₀ / (2π) où γ est le rapport gyromagnétique. Pour ¹H à 11,7 T (500 MHz), ΔE correspond à un photon de radiofréquence (~500 MHz). Une impulsion RF à 90° bascule l'aimantation dans le plan transversal ; sa précession libre (FID) est enregistrée puis transformée en spectre par transformée de Fourier.
Le champ B₀ n'est jamais perçu directement par le noyau : les électrons du nuage périphérique l'écrantent partiellement, ce qui engendre le déplacement chimique δ.
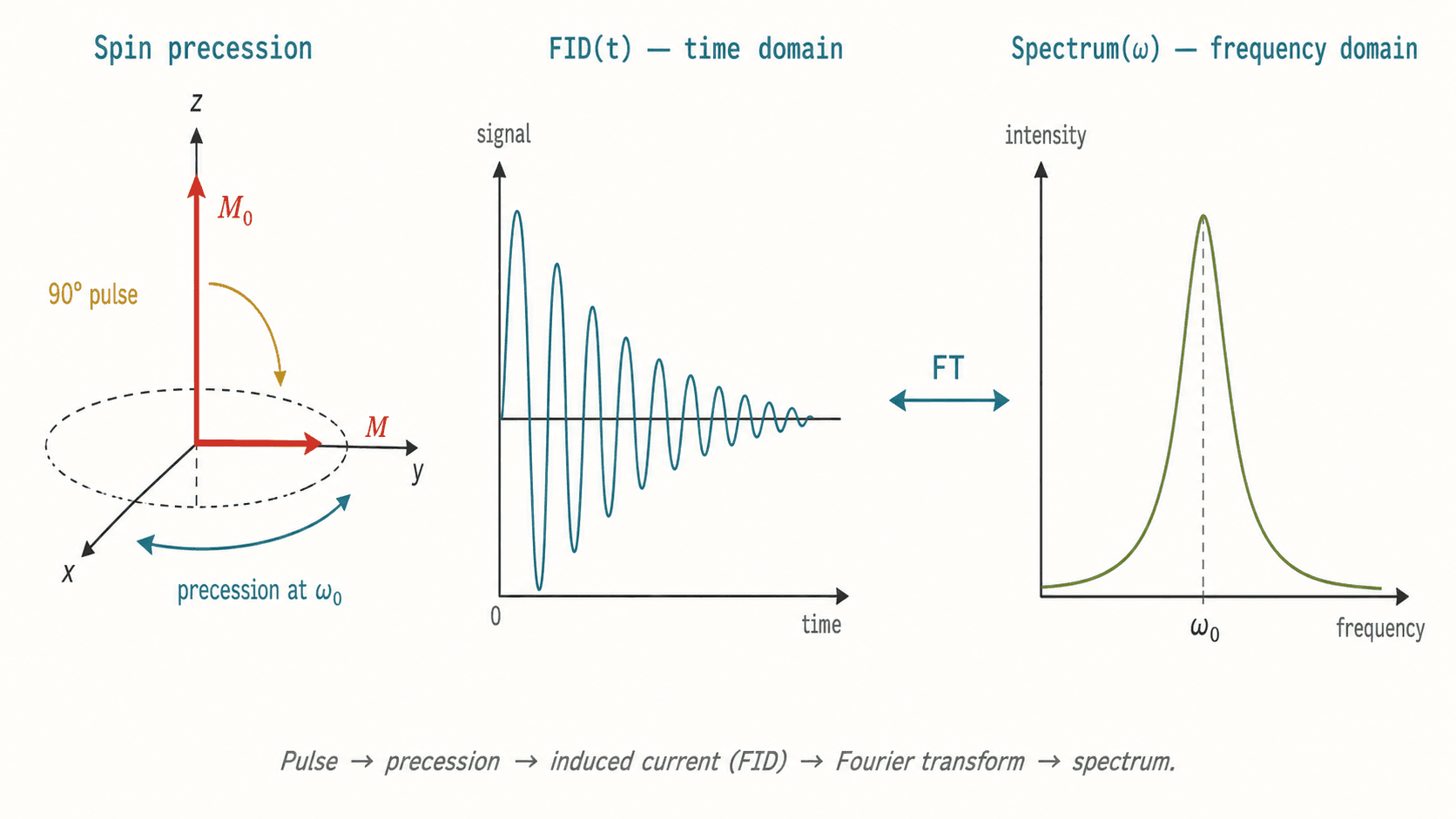
Déplacement chimique δ
Le déplacement chimique est défini par :
δ (ppm) = (ν_échantillon − ν_TMS) / ν_spectrométre × 10⁶
La référence universelle est le tétraméthylsilane (TMS), δ = 0 ppm. Le ppm est sans dimension et indépendant de la fréquence du spectromètre, ce qui rend les spectres comparables entre instruments.
| Environnement ¹H | δ (ppm) |
|---|---|
| TMS | 0 |
| Alkyle (–CH₃, –CH₂–) | 0,8–2,0 |
| Allylique/propargylique | 1,6–2,6 |
| α de C=O | 2,0–2,7 |
| Éther (O–CH) | 3,3–4,5 |
| Vinylique (=CH–) | 4,5–6,5 |
| Aromatique | 6,5–8,5 |
| Aldéhydique (CHO) | 9,0–10,5 |
| Carboxylique (COOH) | 10–12 |
En ¹³C, les déplacements couvrent 0–220 ppm : alkyles (0–50), alcènes/aromatiques (100–160), carbonyles (160–220 ppm).
Couplage scalaire (J)
Les protons géminaux et vicinaux se couplent via les électrons des liaisons (mécanisme scalaire, ³J le plus souvent). Le couplage se manifeste par une division du signal : un proton H_A couplé à n protons équivalents H_X donne un multiplet de (n+1) raies de rapport binomial (règle n+1). Exemples : doublet (d, n=1), triplet (t, n=2), quadruplet (q, n=3).
La constante de couplage ³J (en Hz) dépend de l'angle dièdre φ selon l'équation de Karplus :
³J ≈ A cos²φ − B cos φ + C
Valeurs typiques : ³J_trans (alcène) ≈ 12–18 Hz ; ³J_cis ≈ 6–12 Hz ; ³J (cyclohexane axial-axial, φ ≈ 180°) ≈ 10–13 Hz. L'analyse des constantes J permet de déterminer la stéréochimie.
Le couplage en ¹³C–H est souvent supprimé par découplage des protons largebande (BBD) : on obtient un spectre découplé où chaque carbone donne un singulet. L'expérience DEPT (Distortionless Enhancement by Polarisation Transfer) distingue CH, CH₂ et CH₃ des quaternaires (C absent en DEPT).
Intégration et rapport H
En ¹H RMN, l'aire intégrée d'un signal est directement proportionnelle au nombre de protons qui contribuent à ce signal (à condition d'utiliser un délai de relaxation suffisant, D1 ≥ 5·T₁). L'intégration relative permet de dénombrer les H équivalents par groupe.
Exemple : l'éthanol CH₃CH₂OH donne trois signaux d'intégrales 3:2:1 (CH₃ : CH₂ : OH). L'OH peut être large ou échangeable (disparaît avec D₂O, "D₂O shake").
Stratégie d'attribution complète
L'attribution systématique d'un spectre ¹H/¹³C obéit à la séquence :
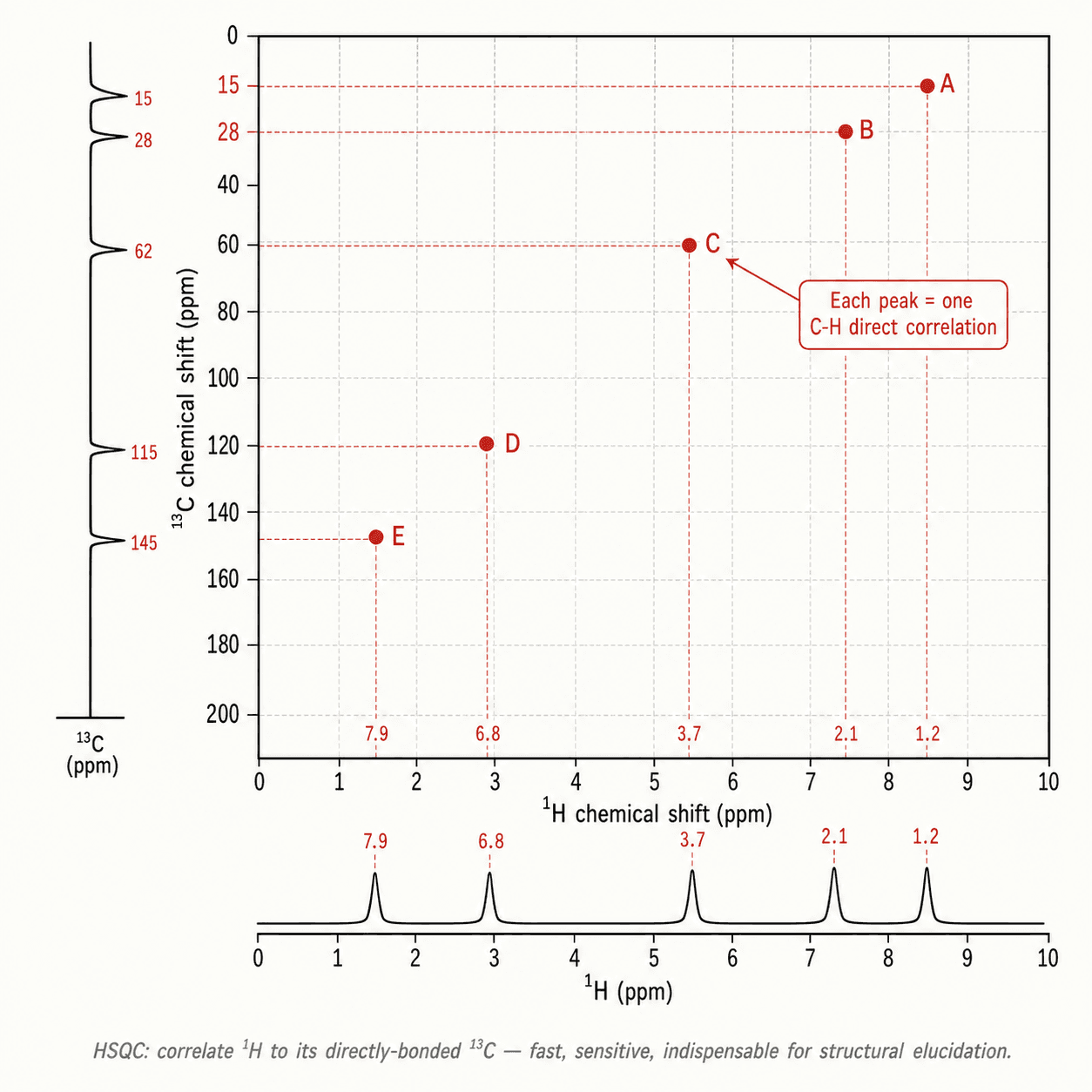
1. Formule brute et degré d'insaturation (DI = (2C + 2 + N − H) / 2). Un DI élevé oriente vers aromatique ou carbonyle. 2. ¹³C DEPT : inventorier les types de carbone (CH, CH₂, CH₃, C quat.). 3. ¹H δ et intégrations : identifier les groupements caractéristiques. 4. Multiplicités et J : chaîner les fragments (CH₃ triplet → CH₂ voisin). 5. Corrélations 2D (COSY, HSQC, HMBC) pour les molécules complexes : COSY relie H–H vicinaux ; HSQC relie H directement à C ; HMBC relie H à C à 2–3 liaisons.
Aspects pratiques et solvants deutériés
Les spectres sont enregistrés en solvants deutériés pour éviter le signal massif du solvant protoné. Les solvants courants et leur signal résiduel : CDCl₃ (δ_H = 7,26 ; δ_C = 77,0), DMSO-d₆ (δ_H = 2,50 ; δ_C = 39,5), D₂O (δ_H = 4,79). Le signal résiduel est souvent utilisé comme référence interne quand le TMS n'est pas ajouté. La concentration usuelle est 10–50 mg dans 0,6 mL de solvant ; un excès de soluté dégrade la résolution par viscosité.